![]() Follow me on twitter @BarryDigby
Follow me on twitter @BarryDigby
![]() Follow me on twitter @BarryDigby
Follow me on twitter @BarryDigby

We will use nextflow to construct a script capable of performing read QC, adapter/read filtering and subsequent QC on the trimmed reads.
Jump to:
I will show you how to construct the processes, leaving you to stitch them together to make a single script.
In nextflow, parameters are variables that can be passed to the script. You can hard-code them in the script, or pass them to the script via the command line. Whichever method you choose, the parameter must be declared in the script i.e params.reads = "/path/*glob" or params.reads = null
Save the below script as test.nf and run nextflow run test.nf
#!/usr/bin/env nextflow
// Parameters
params.reads = "/data/MSc/2020/MA5112/week_1/raw_data/*_r{1,2}.fastq.gz"
// Initialise Channel
reads_ch = Channel.fromFilePairs(params.reads)
reads_ch.view()
Pass a different FASTQ pair to the script via the command line by calling:
nextflow run test.nf --reads "/data/MSc/2020/MA5112/Variant_Calling/reads/*_r{1,2}.fastq.gz"
In the previous script test.nf, the output printed to the terminal showed you the structure of the files in the channel reads_ch. We used fromFilePairs(), which is specifically designed for paired end sequencing data. It works by creating a tuple in which the first element is the grouping key of the matching pair and the second element is the list of files (sorted in lexicographical order).
Let’s practice accessing the contents of these tuples in a process.
Save the below script as test1.nf and run nextflow run test1.nf
#!/usr/bin/env nextflow
// Parameters
params.outdir = "./"
params.reads = "/data/MSc/2020/MA5112/week_1/raw_data/*_r{1,2}.fastq.gz"
// Initialise Channel
reads_ch = Channel.fromFilePairs(params.reads)
process test{
echo true
input:
tuple val(base), file(reads) from reads_ch
output:
stdout to out
script:
"""
echo "$base"
echo ""
echo "$reads"
"""
}
During the tutorial we ran into errors with pulling the docker image from Dockerhub via the nextflow config file. I also ran into a .nextflow/history.lock (no such file or directory) error when attempting to run nextflow in my directory.
Firstly we will fix the permissions error:
nextflow run commands) there is a hidden folder called .nextflow/. Change the permissions to this directory and delete it:chmod -R 777 .nextflow/
rm -rf .nextflow/
chmod -R 777 work/
rm -rf work/
Next we will examine the ~/.nextflow/config file:
process {
beforeScript = 'module load singularity'
containerOptions = '-B /data/'
container = 'barryd237/week1:test'
executor='slurm'
queue='normal'
clusterOptions = '-n 1'
}
singularity.enabled = true
singularity.autoMounts = true
singularity{
cacheDir = '/data/MSc/2021/container_cache'
}
(This confguration file worked for the script quality_control.nf given below.)
We are telling the config file to pull the image from barryd237/week1:test and to store it under /data/MSc/2021/container_cache.
When you run the command for the first time, nextflow will output this message:
Pulling Singularity image docker://barryd237/week1:test [cache /data/MSc/2021/container_cache/barryd237-week1-test.img].
As I have already pulled the container to this directory, this message should not be printed and the processes will run automatically.
If for some reason nextflow complains about container permissions (or you want to test this out yourself), create a container_cache directory in your own directory, and update the .nextflow/config file accordingly i.e : cacheDir = '/data/MSc/2021/username/container_cache'.
Finally, if nextflow complains that it cannot publish the files to the QC/ directory, I am willing to bet that the QC/ directory has unknown permissions:
?????????? ? ? ? ? hcc1395_normal_rep1_r1_fastqc.html
?????????? ? ? ? ? hcc1395_normal_rep1_r1_fastqc.zip
?????????? ? ? ? ? hcc1395_normal_rep1_r2_fastqc.html
?????????? ? ? ? ? hcc1395_normal_rep1_r2_fastqc.zip
?????????? ? ? ? ? hcc1395_normal_rep2_r1_fastqc.html
?????????? ? ? ? ? hcc1395_normal_rep2_r1_fastqc.zip
To fix this, change the permissions to the directory and delete it:
chmod -R 777 QC/
rm -rf QC/
Note: this might affect other publishDir directories, check if they have incompatible user permissions (?) and delete them in the same manner given above.
In nextflow files are passed to each process via channels.
In the previous example we created the channel reads_ch by specifying Channel.fromFilePairs(). We will use the read_ch to provide the FastQC process the raw data, collect the files prodcued from FastQC and pass them to MultiQC, demonstrating inputs and output usage in processes.
N.B: Channels can only be used once!
#!/usr/bin/env nextflow
// Parameters
params.outdir = "./"
params.reads = "/data/MSc/2020/MA5112/week_1/raw_data/*_r{1,2}.fastq.gz"
// Initialise Channel
reads_ch = Channel.fromFilePairs(params.reads)
process FastQC {
publishDir "${params.outdir}/QC/raw", mode:'copy'
input:
tuple val(key), file(reads) from reads_ch
output:
file("*.{html,zip}") into fastqc_ch
script:
"""
fastqc -q $reads
"""
}
process MultiQC {
publishDir "${params.outdir}/QC/raw", mode:'copy'
input:
file(htmls) from fastqc_ch.collect()
output:
file("*multiqc_report.html") into multiqc_report_ch
script:
"""
multiqc .
"""
}
Save the script as quality_control.nf.
To run the script on Lugh we will add additional parameters to the command line:
```
nextflow -bg -q run quality_control.nf \
-with-singularity /data/MSc/2020/MA5112/week_1/container/week1.img
```
-bg Run the script in the background. Nextflow will parse the Configuration file located at ~/.nextflow/config to determine the resource usage for SLURM.-q Quiet, do not print stdout to console.-with-singularity Use container to execute the script. Provide the path to the container.Run this line of code yourself, and type squeue -u mscstudent on Lugh to view the submitted jobs.
UPDATE You should be able to run the above script by simply running:
```
nextflow -bg run quality_control.nf
```
The container + container_cache specified in the config file should now work. This is also applicable to the exercise, you should not have to specify the container via the command line each time.
I have shown you how to intitialise a raw read channel for downstream use with FastQC and MultiQC.
In the exercise below I have started a nextflow script to read in the raw reads and perform adapter trimming and read filtering using bbduk.sh. Finish the script by using the trimmed reads channel, passing it to FastQC, and collecting the outputs of FastQC as inputs to Multiqc.
If this tasks seems daunting, break the script into processes. First, include only the first process in the script, and add
trimmed_reads_ch.view()after the process block to check the outputs. Keep adding processes iteratively - this is how I develop nextflow pipelines, writing multiple processes in one run is asking for trouble :)
Save as trim_qc.nf and run in your own directory:
nextflow -bg -q run trim_qc.nf \
--outdir $(pwd) \
-with-singularity /data/MSc/2020/MA5112/week_1/container/week1.img
or
nextflow -bg run trim_qc.nf --outdir $(pwd)
#!/usr/bin/env nextflow
// Parameters
params.outdir = "./"
params.reads = "/data/MSc/2020/MA5112/week_1/raw_data/*_r{1,2}.fastq.gz"
params.adapters = "/data/MSc/2020/MA5112/week_1/assets/adapters.fa"
// Initialise Channel
reads_ch = Channel.fromFilePairs(params.reads)
process Trim {
label 'BBDUK'
publishDir "${params.outdir}/trimmed_reads", mode:'copy', pattern: "*.fq.gz"
publishDir "${params.outdir}/BBDUK_stats", mode:'copy', pattern: "*.stats.txt"
input:
tuple val(key), file(reads) from reads_ch
path(adapters) from params.adapters
output:
tuple val(key), file("*.fq.gz") into trimmed_reads_ch
file("*.stats.txt") into trimmed_stats_ch
script:
"""
bbduk.sh -Xmx4g \
in1=${reads[0]} \
in2=${reads[1]} \
out1=${key}_r1.fq.gz \
out2=${key}_r2.fq.gz \
ref=$adapters \
minlen=30 \
ktrim=r \
k=12 \
qtrim=r \
trimq=20 \
stats=${key}.stats.txt
"""
}
process FastQC {
label 'FastQC'
publishDir "${params.outdir}/QC/trimmed", mode:'copy'
input:
output:
script:
"""
fastqc -q $reads
"""
}
process MultiQC {
label 'MultiQC'
publishDir "${params.outdir}/QC/trimmed", mode:'copy'
input:
output:
script:
"""
multiqc .
"""
}
nextflow -bg -q trim_qc.nf -with-singularity /<path to>/week1.img.Move the multiqc_report.html file under QC/trimmed to your local machine using scp:
# This pushes the file to bactsrv
scp multiqc_report.html username@bactsrv.nuigalway.ie:/home/username/
Now move to your local machine and run:
# Pull the file from bactsrv to current dir ('.')
scp username@bactsrv.nuigalway.ie:/home/mscstudent/multiqc_report.html .
Check that your MultiQC outputs are the same as the screenshots below:
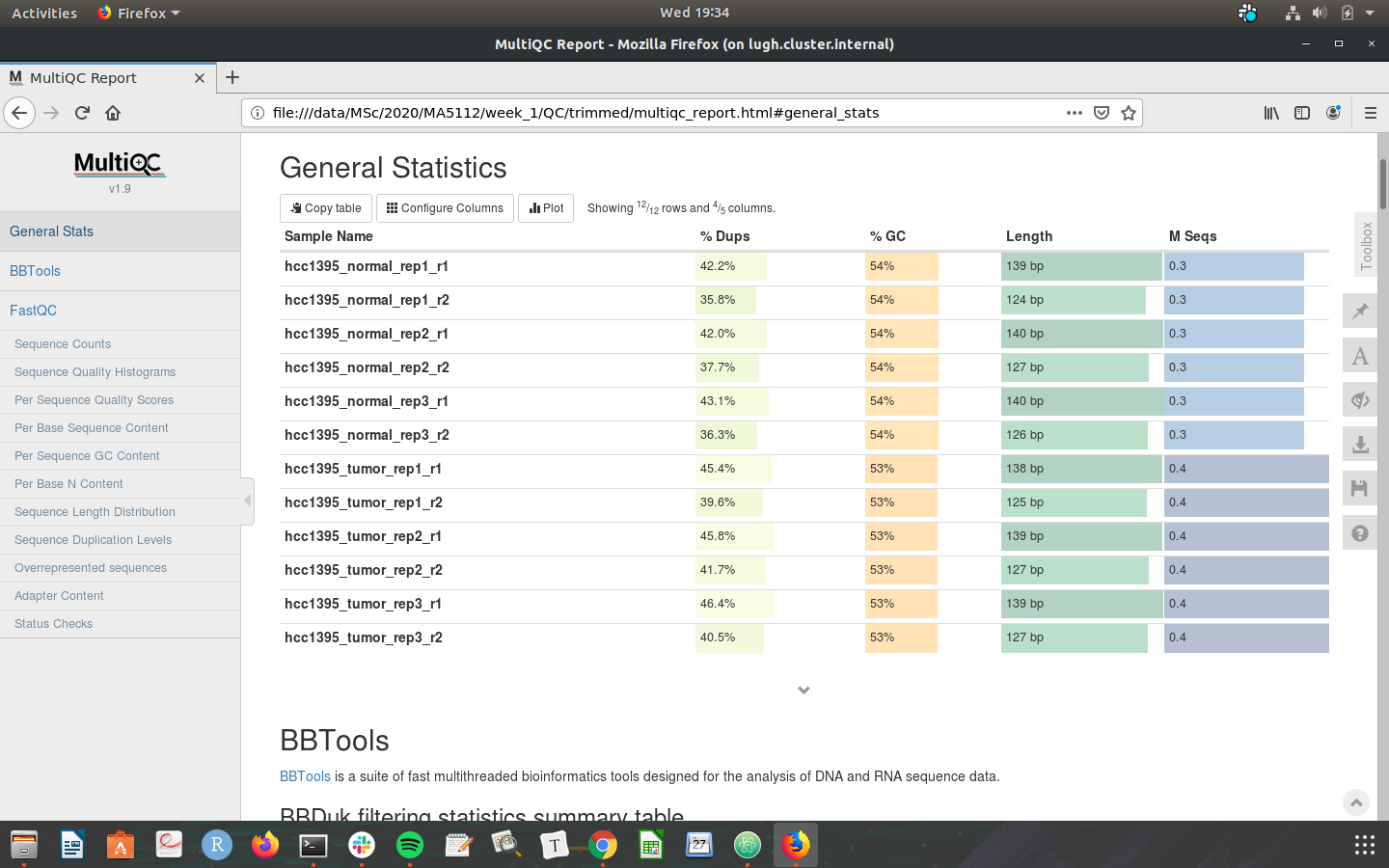
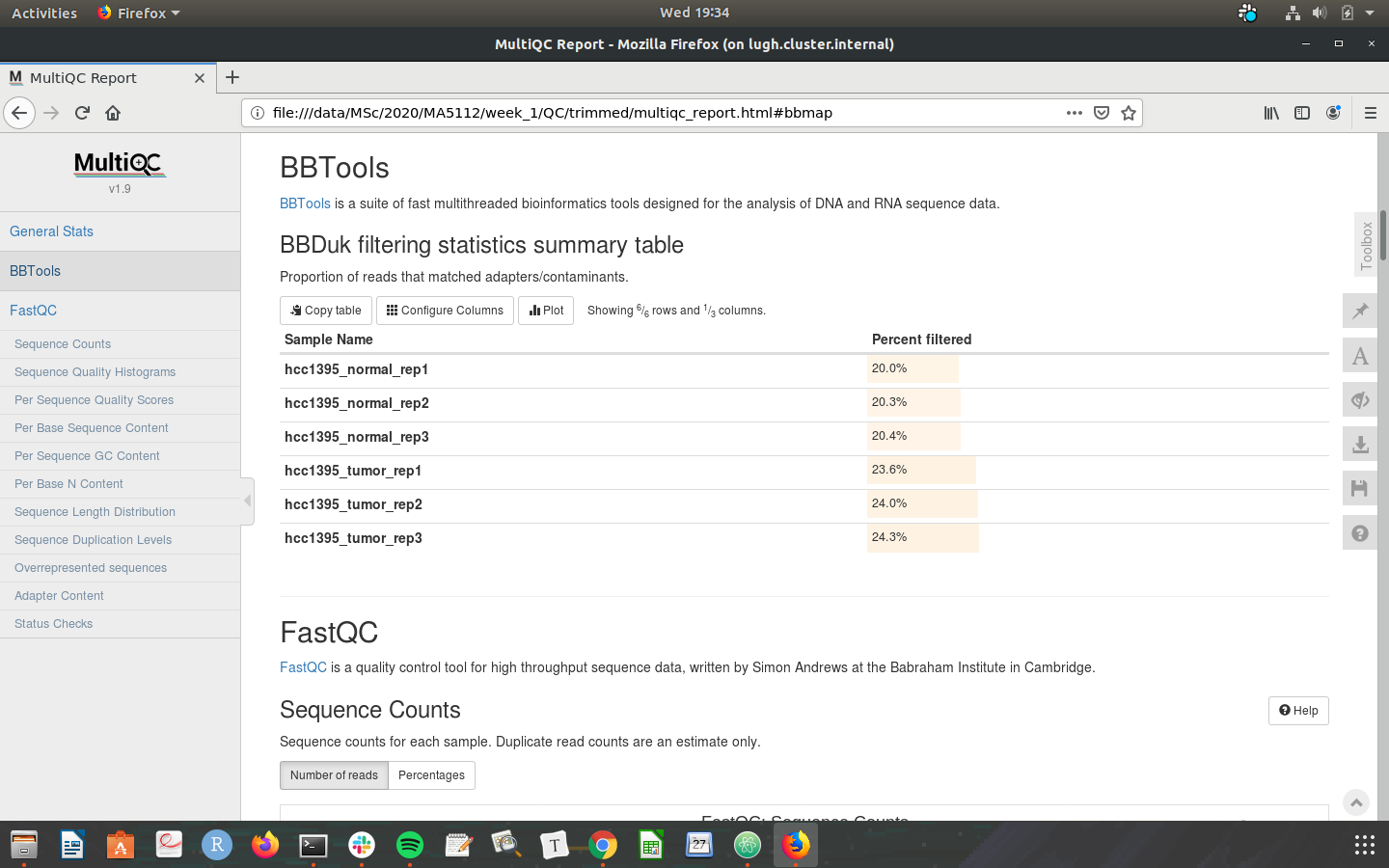
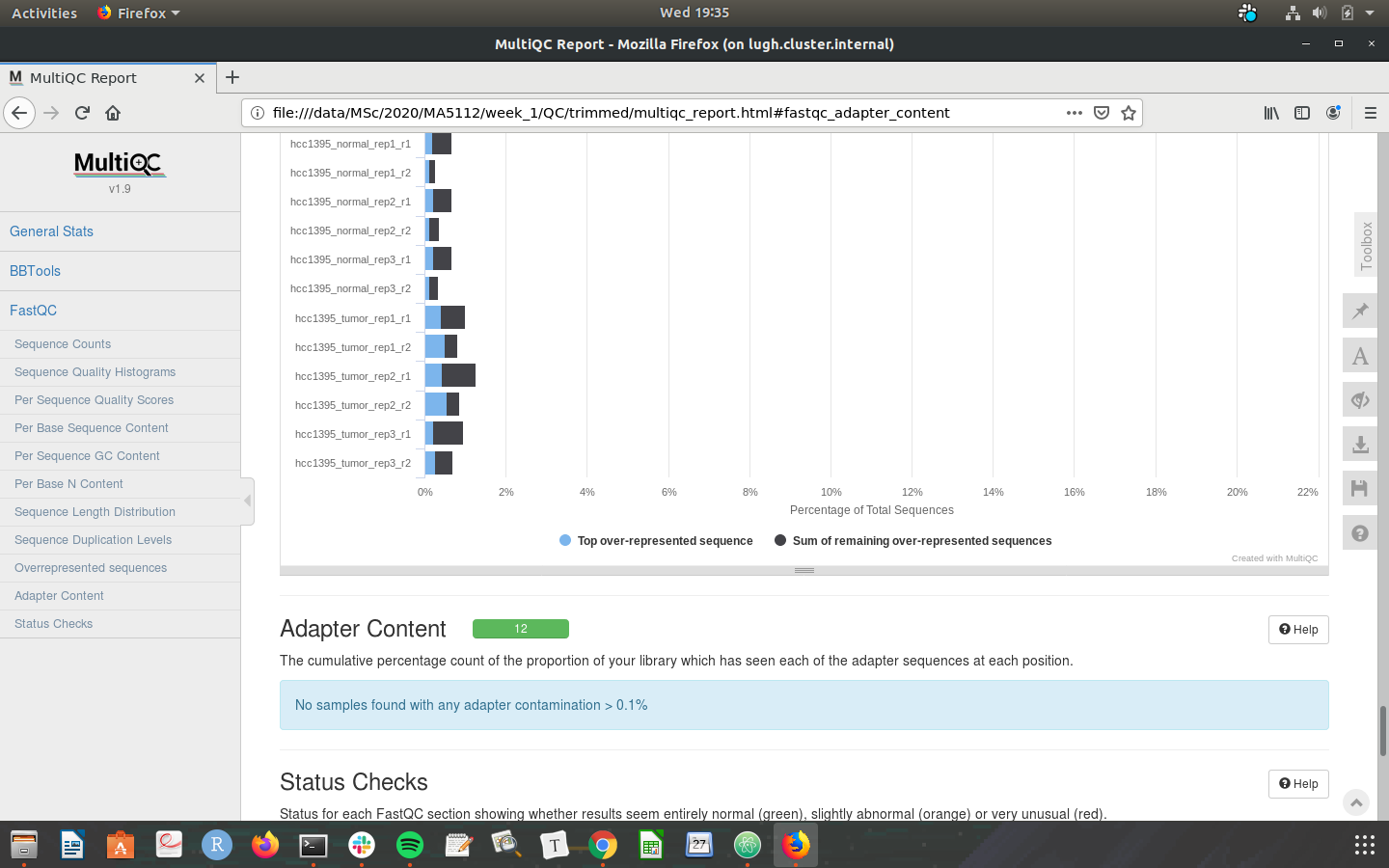
Congratulations on making it through the week 1 tutorial. Please do not hesistate to post in the slack channel if you need help - preferably to a public channel!
As the tutorials progress I expect you to help each other with troubleshooting or I will go AFK..
Files used for the tutorial are available at the following link.
Barry